Divide-conquer HF by Kobayashi and Nakai¶
Case 1: Helium chains¶
The simplest case of closed-shell non-interacting systems assembled into a single model. The Helium atoms are arranged into chains with 6A spacing.
Convergence¶
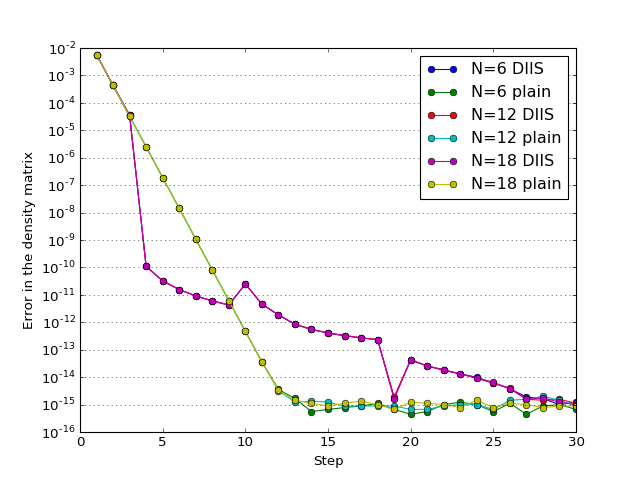
The study of how the error in density matrix elements decreases with iterations; \(N\) is the number of Helium atoms in the model.
(Source code, png, hires.png, pdf)
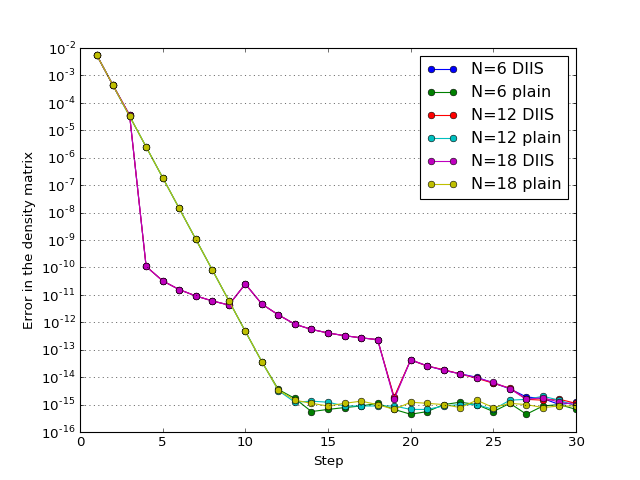{kind=link}
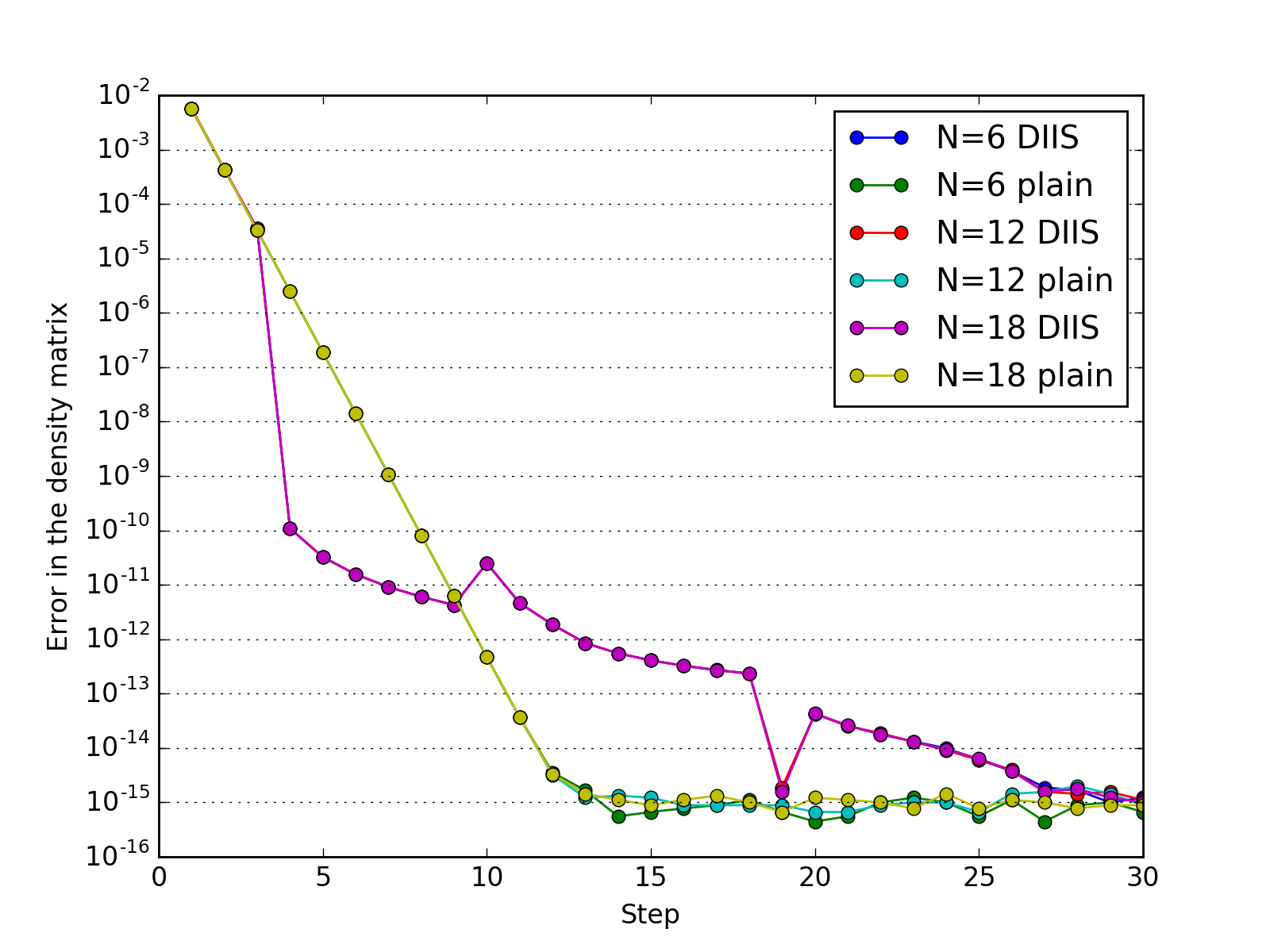{kind=link}
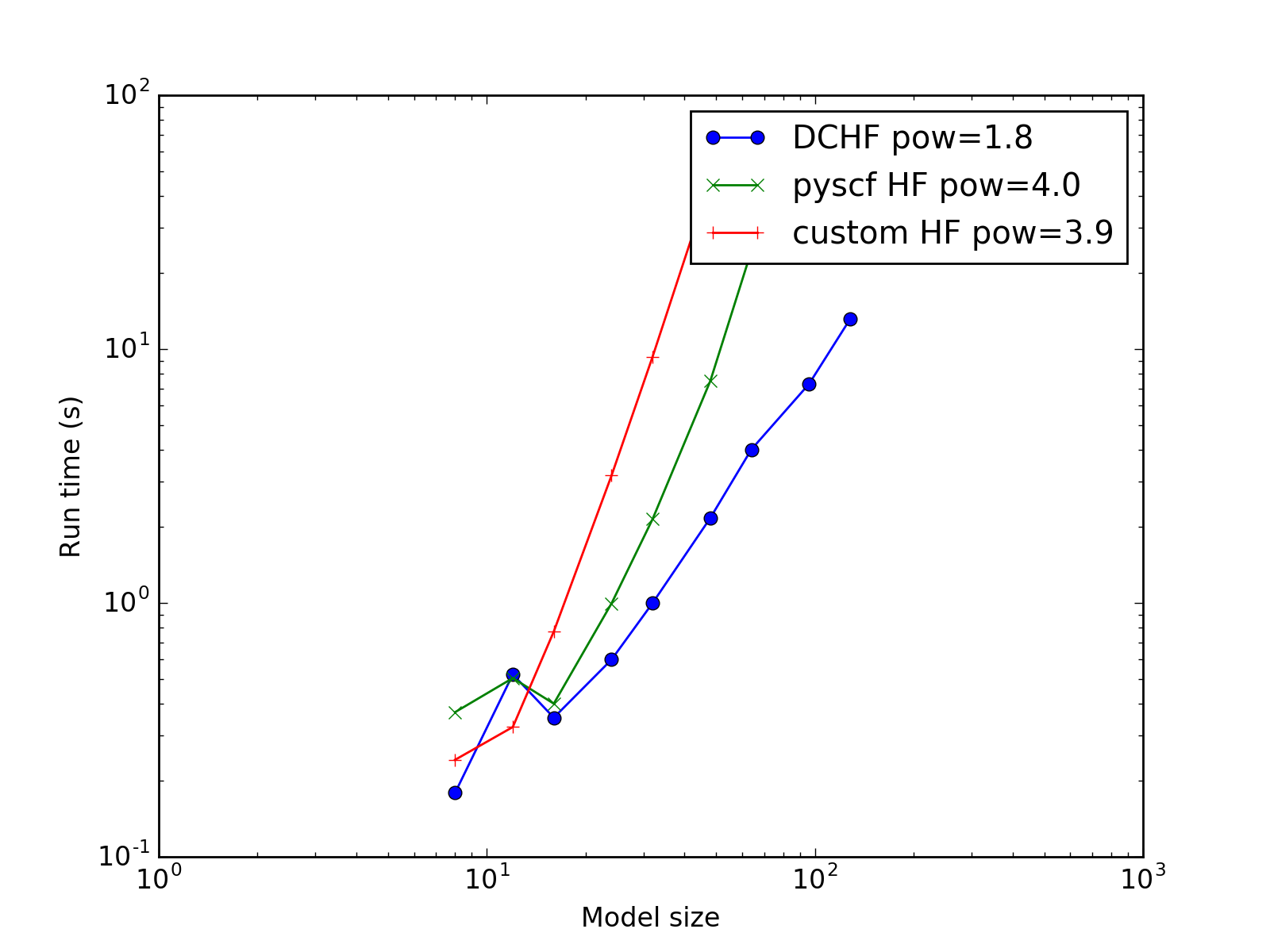
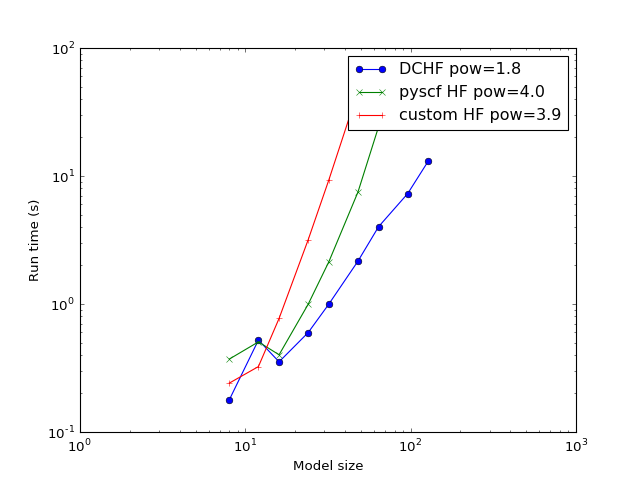
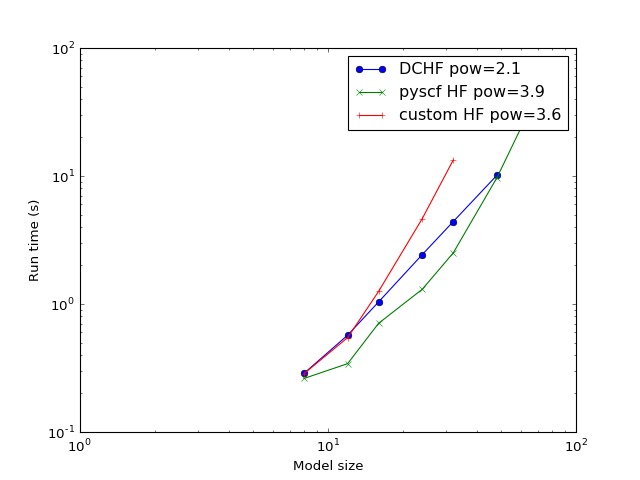
Complexity¶
The study of how the calculation time scales with the model increase. The expected problem complexity in conventional HF is \(O(N^4)\) while this implementation of DC-HF should scale as \(O(N^2)\) where \(N\) is the number of atoms in the system.
(Source code, png, hires.png, pdf)
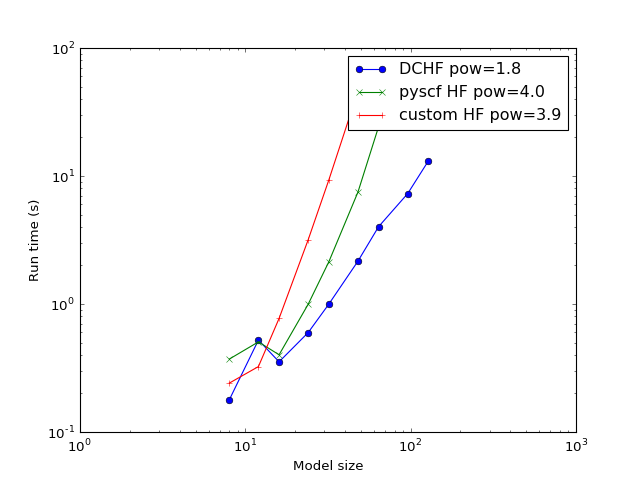{kind=link}
{kind=link}
Case 2: Hydrogen dimer chains¶
Study of 1D dimerized chains of hydrogen atoms where the minimal distance between nearest neighbours is 1.4A and the maximal distance is 2.3A.

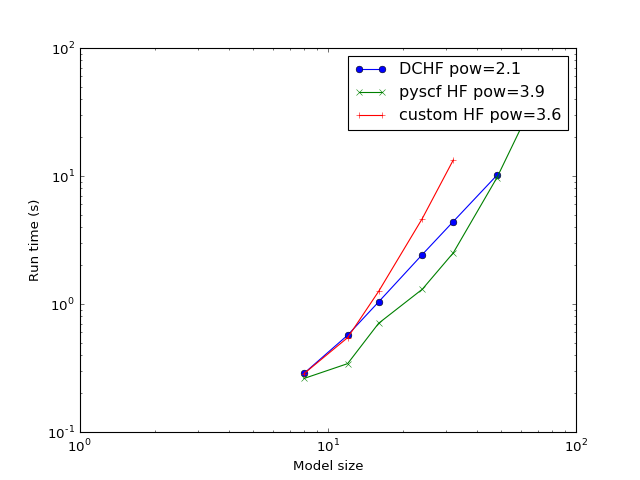
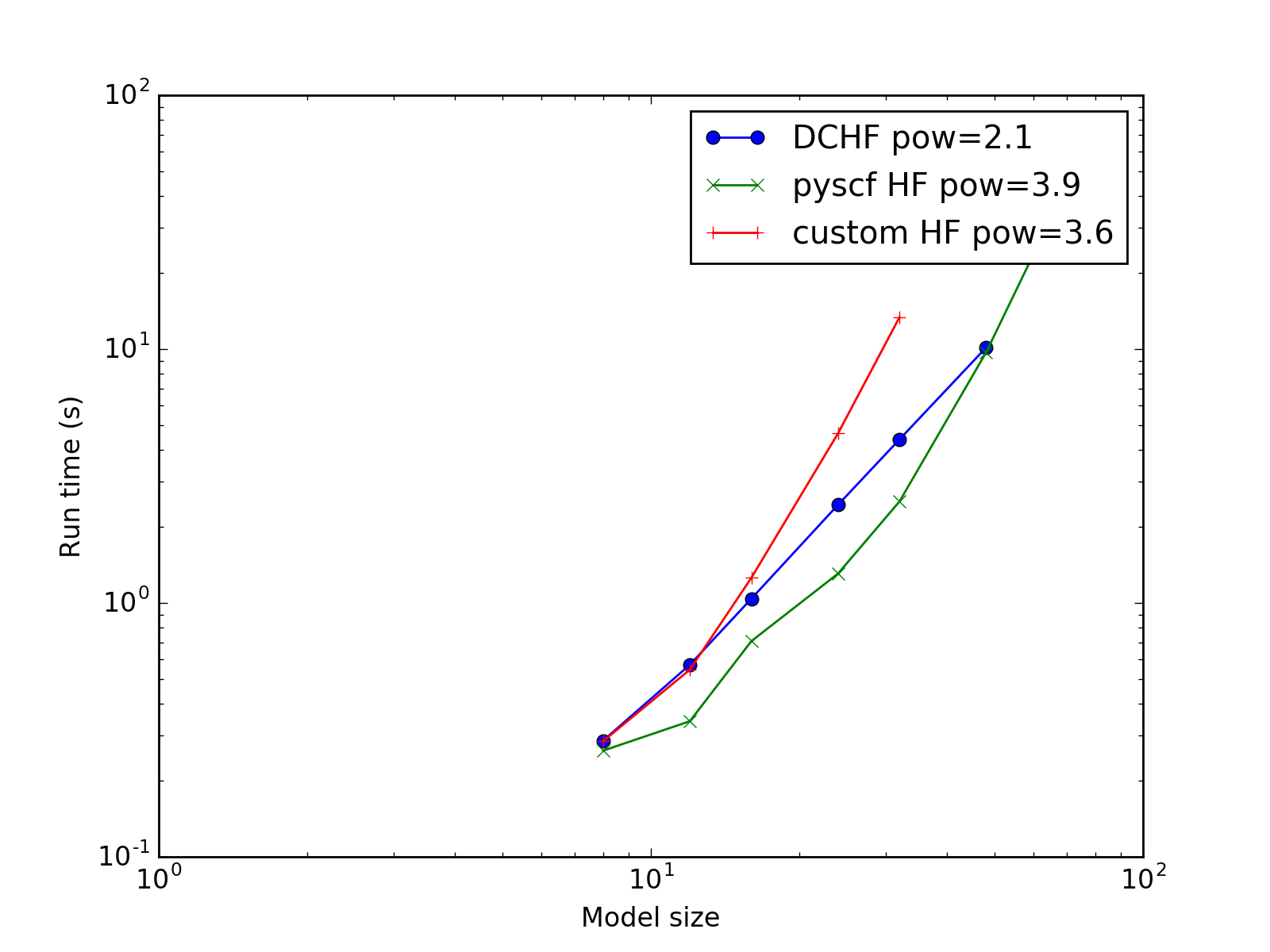
Complexity¶
The subject model is split into domains with 4 atoms in the core region and 2 atoms in the buffer region at both sides of the domain (the 4(2) configuration).

The complexity is expected to be reduced from \(O(N^4)\) of conventional HF to \(O(N^2)\) for this implementation.
(Source code, png, hires.png, pdf)
{kind=link}
{kind=link}
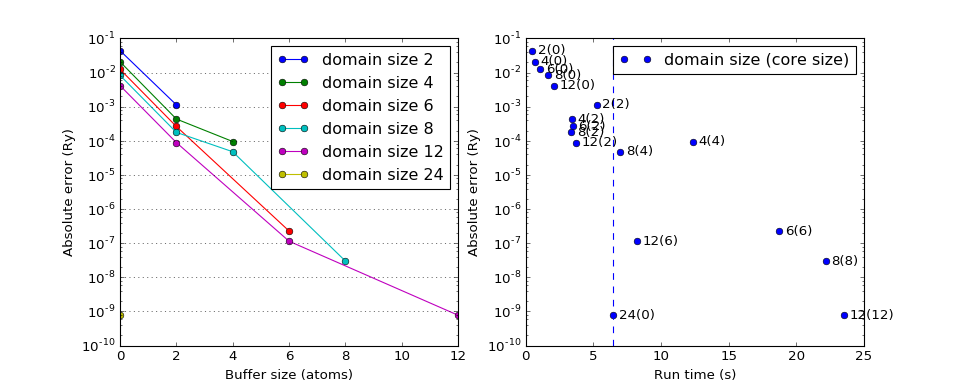
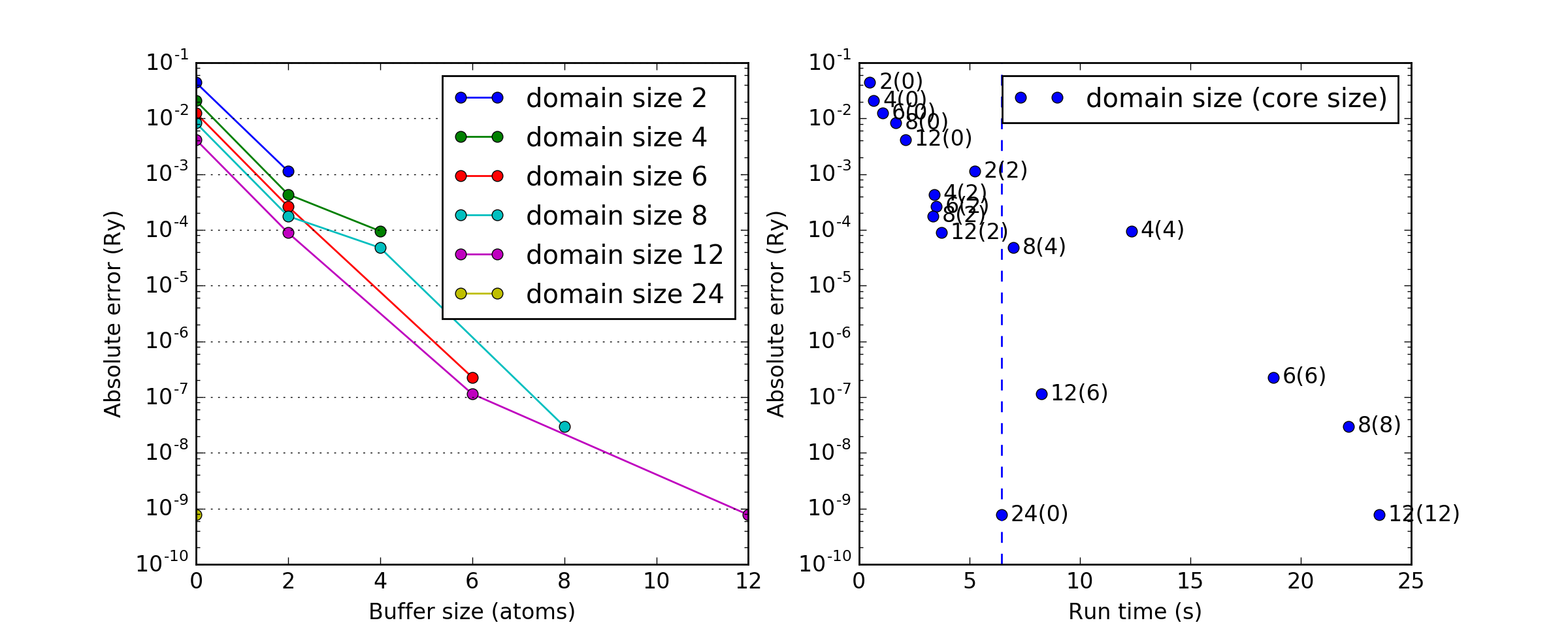
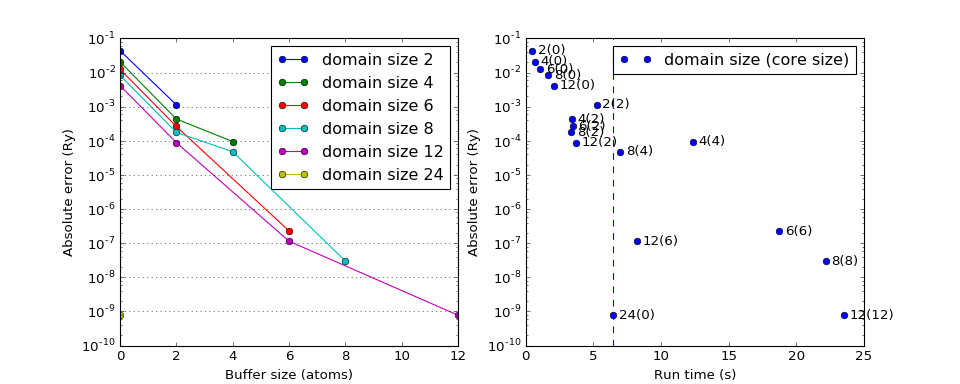
Domain size dependence¶
Errors in HF energies (compared to conventional HF) as a function of the domain size, buffer size and run time. The size of the chain considered is \(N=24\).
(Source code, png, hires.png, pdf)
{kind=link}
{kind=link}
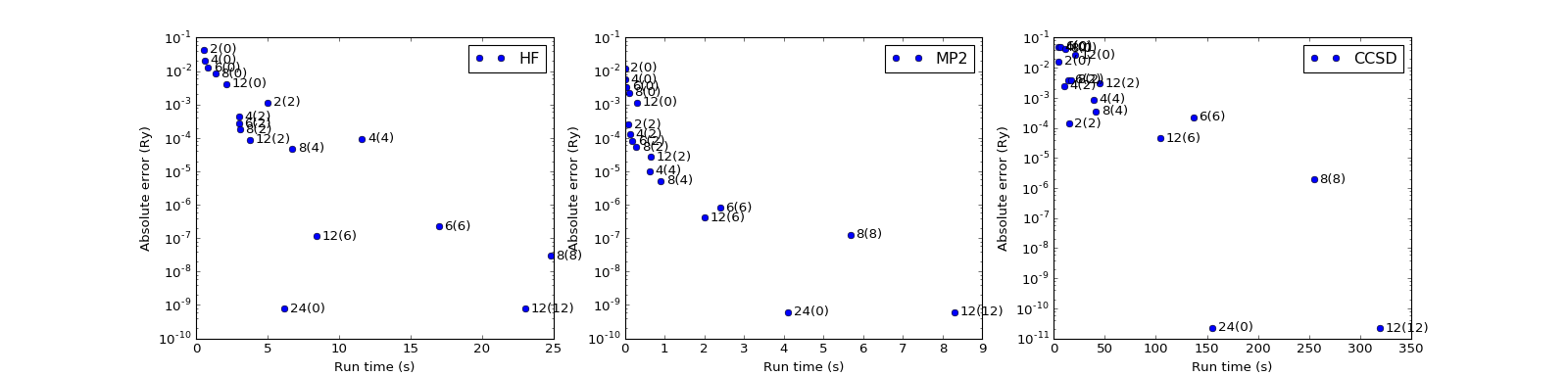
MP2 and CCSD energy corrections¶
Errors in total energies \(E_\mathrm{HF}\) (HF) and \(E_\mathrm{corr}\) (MP2, CCSD; compared to conventional approaches) as a function of the domain size, buffer size and the run time. The chain size is \(N=24\).
(Source code, png, hires.png, pdf)
{kind=link}
{kind=link}
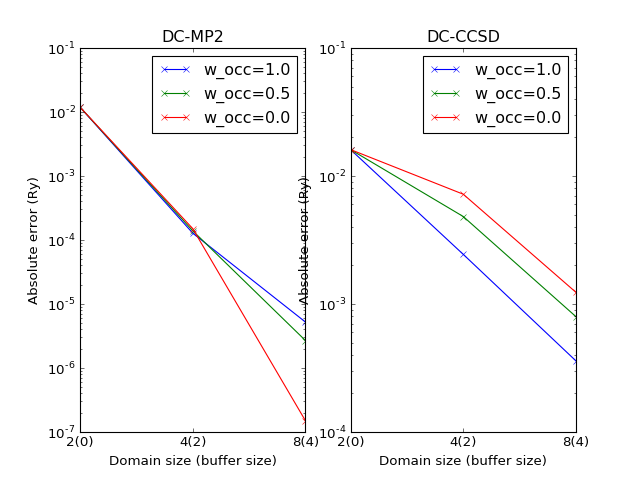
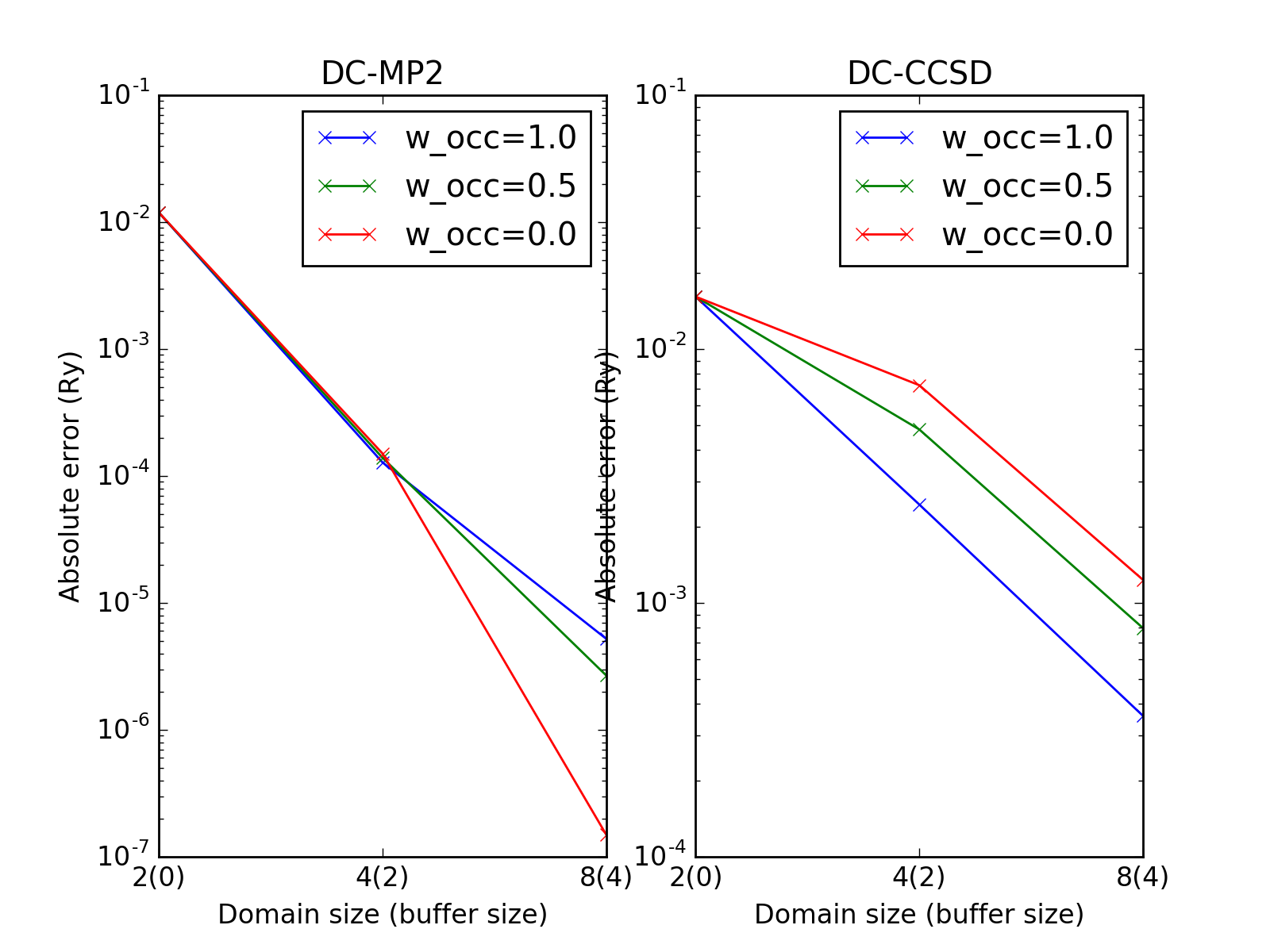
The errors slightly depend on the theory parameter \(w_\mathrm{occ}\) as shown on the following plot.
(Source code, png, hires.png, pdf)
{kind=link}
{kind=link}
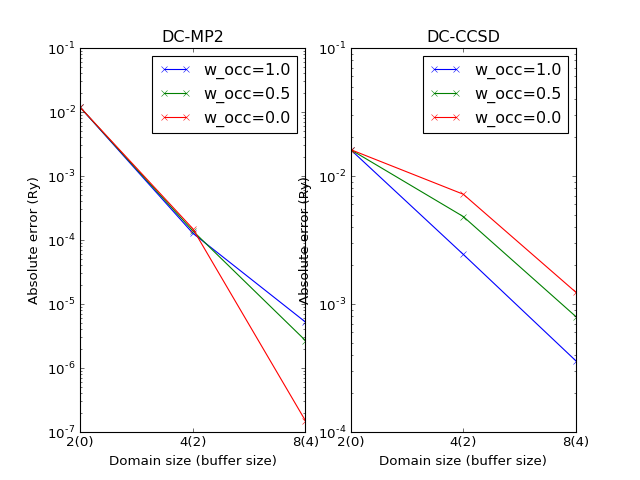
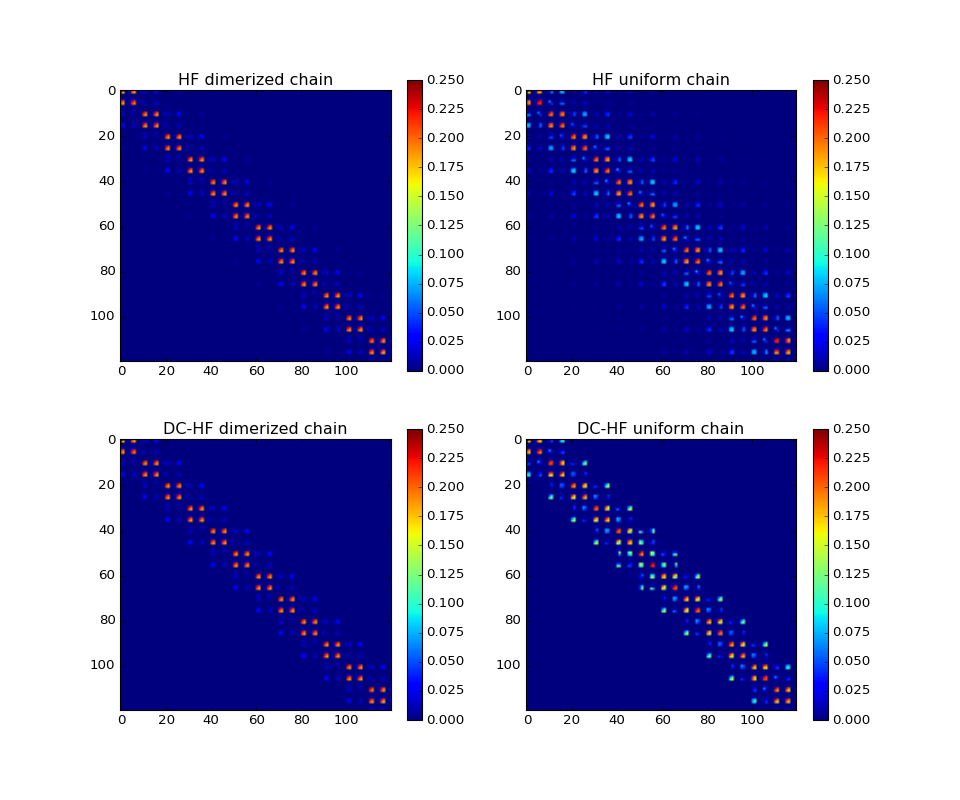
Difficult cases¶
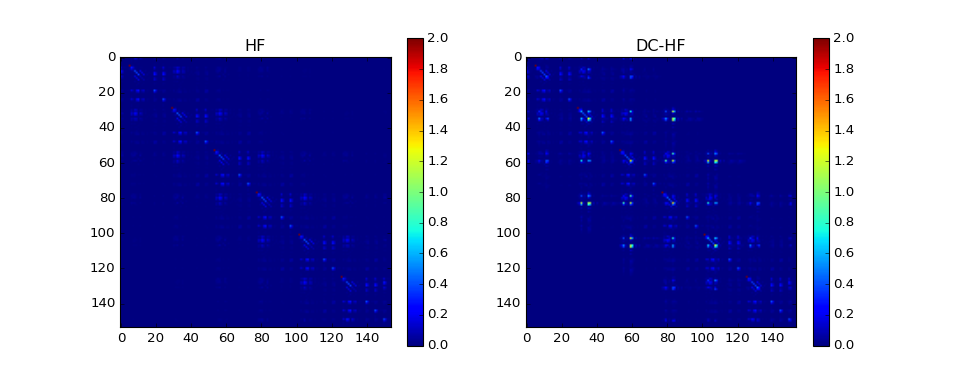
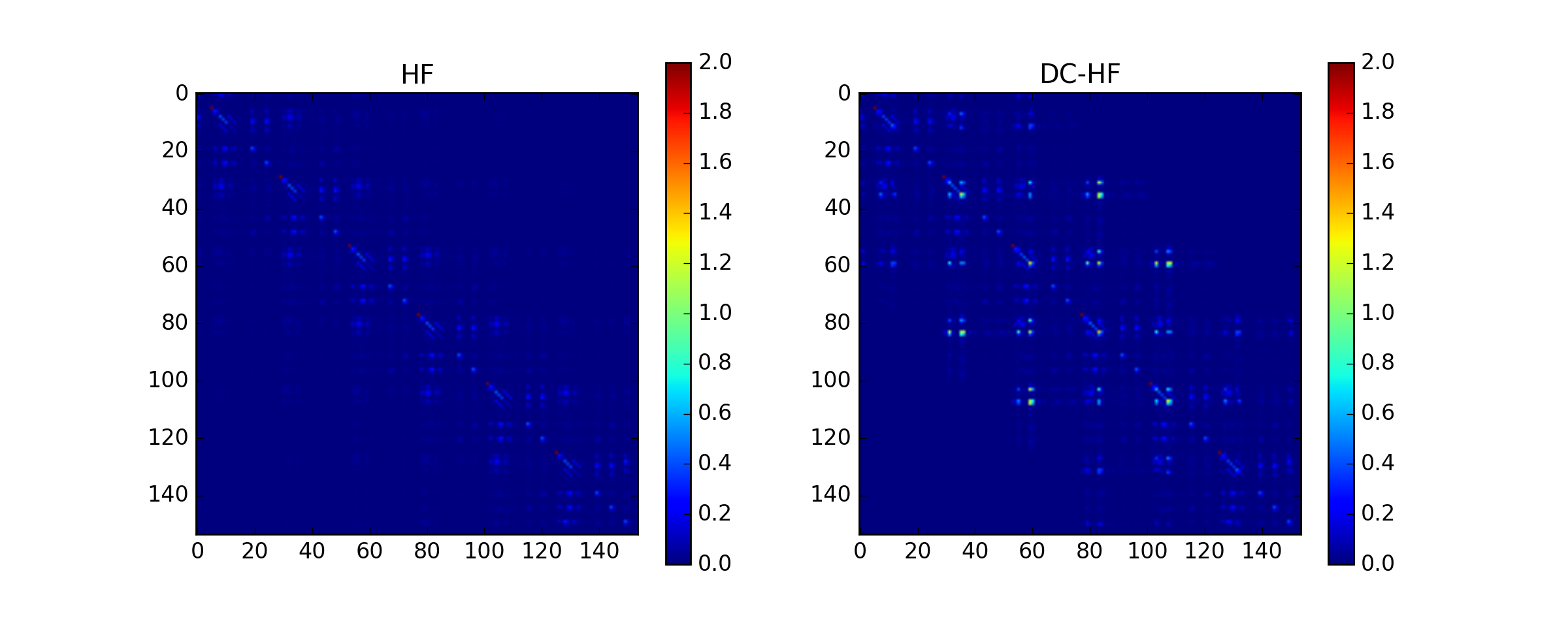
The DC-HF essentially assumes locality of molecular orbitals. The more molecular orbitals extend, the larger error will be. The dimerization in hydrogen chains prevents delocalization of orbitals. Once the spacing between nearest hydrogens becomes equal, the orbitals become delocalized and the approximation fails. The following two plots demonstrate absolute values of density matrixes (color) obtained from a conventional HF calculation and DC-HF for both dimerized and uniform chains.
(Source code, png, hires.png, pdf)
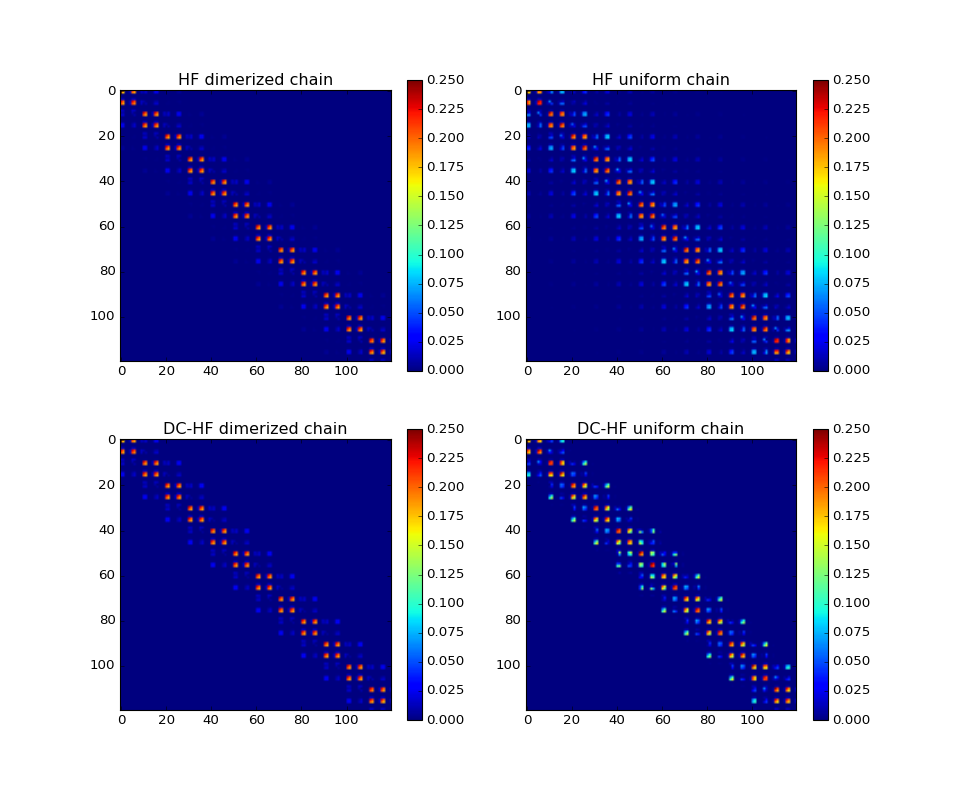{kind=link}
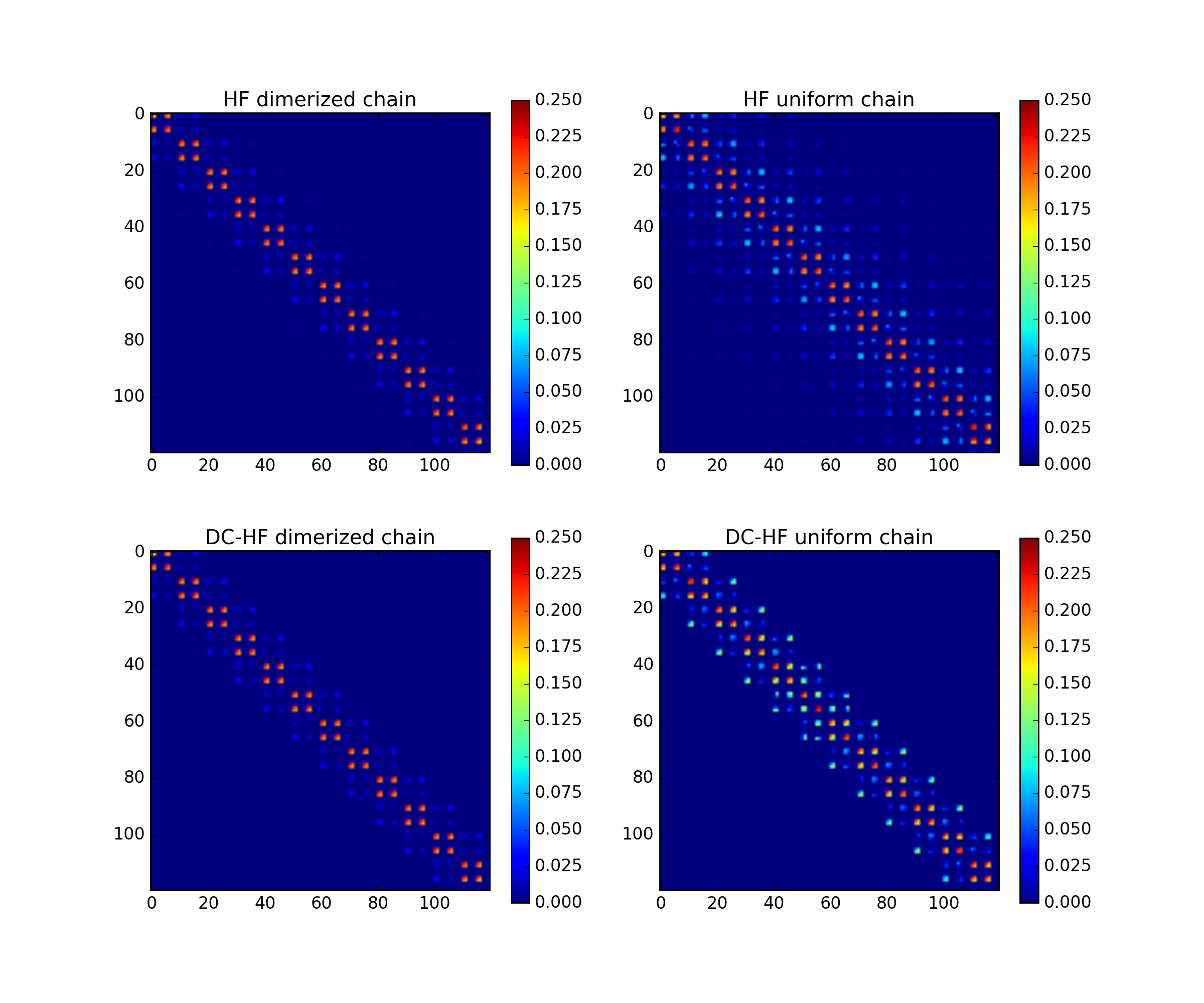{kind=link}
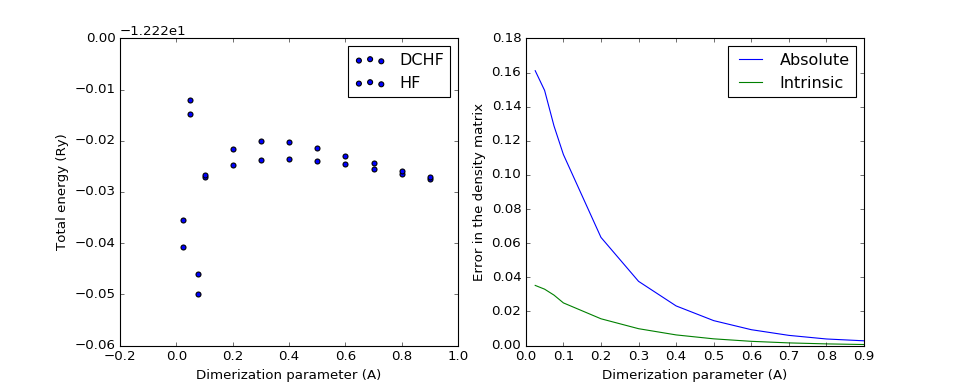
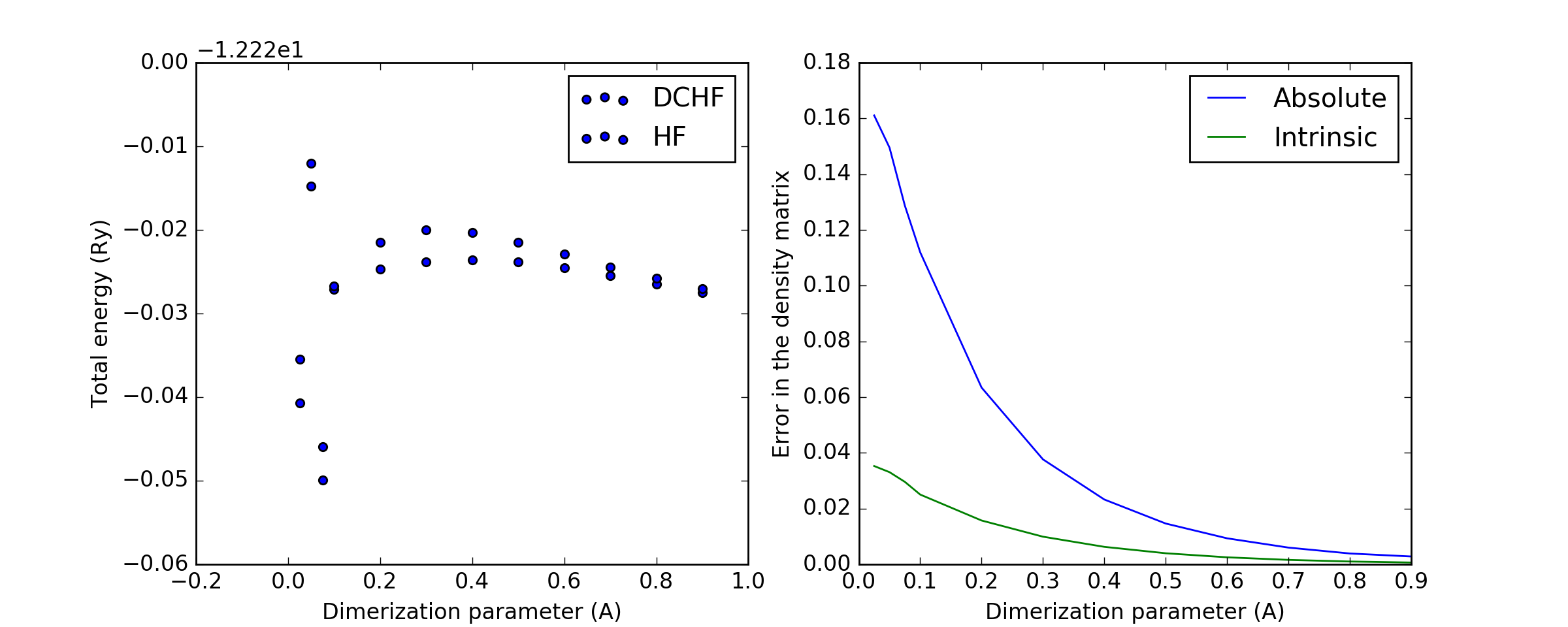
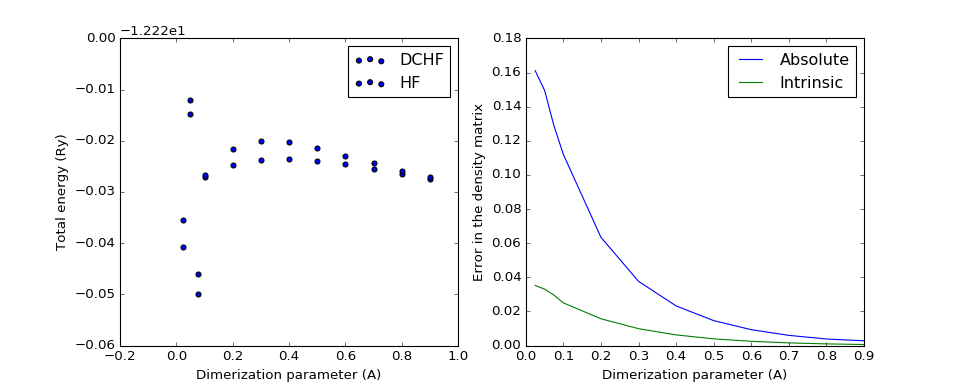
The following two plots demonstrate how the total energy and the errors in DC-HF density matrix behave on the change of dimerization parameter, the difference between the maximal and the minimal distance between nearest neighbours in the chain. The minimal distance is kept constant at 1.4A.
(Source code, png, hires.png, pdf)
{kind=link}
{kind=link}
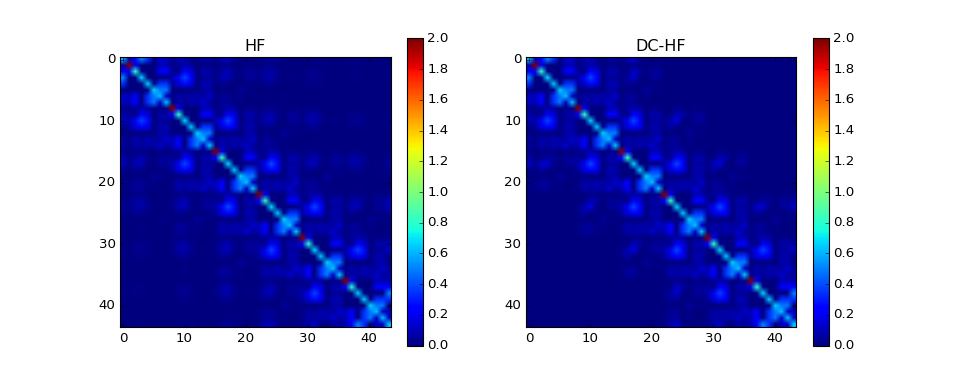
Case 3: Linear alkanes¶
The benchmark model is \(\mathrm{C_{6} H_{14}}\) split into 6 clusters.

Basis set: cc-pvdz¶
The density matrix comparison:
(Source code, png, hires.png, pdf)
{kind=link}
{kind=link}
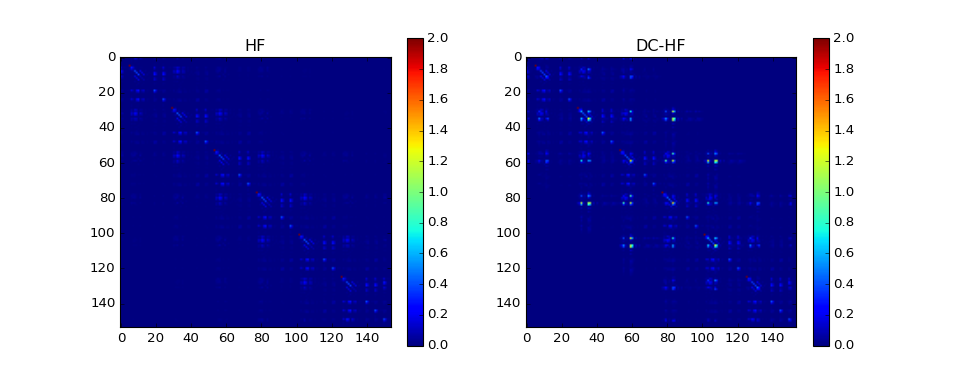
DM error: 1.27165943285
DM intrinsic error: 0.00862713587552
Energy diff: 0.769603364653
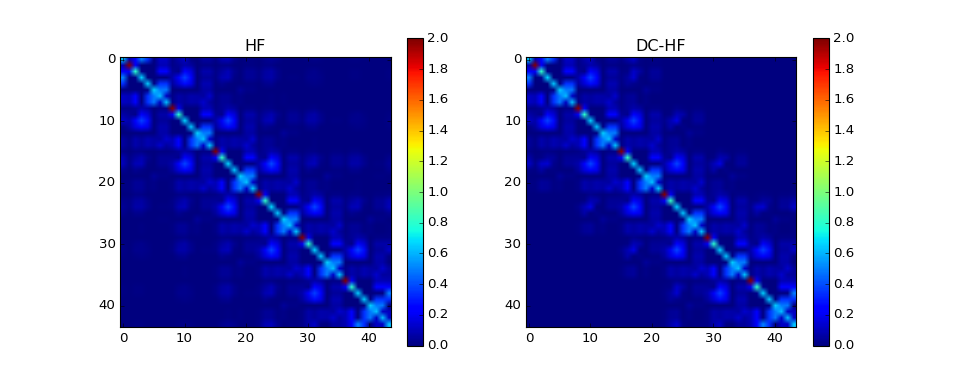
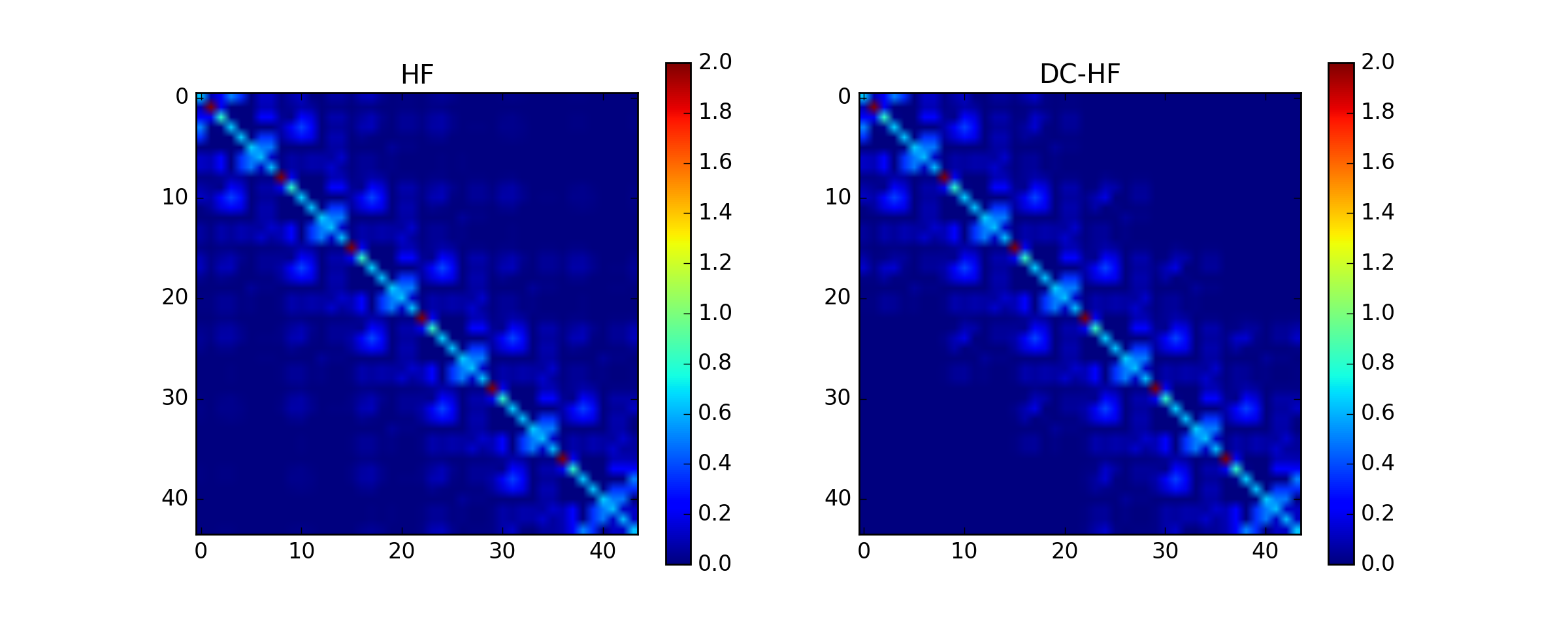
Basis set: sto3g¶
The density matrix comparison:
(Source code, png, hires.png, pdf)
{kind=link}
{kind=link}
DM error: 0.0747824565832
DM intrinsic error: 0.0108856898115
Energy diff: 0.0193028866541